Molecular Docking
About this tutorial
This tutorial demonstrates molecular docking of a small molecule drug to a protein target. We will dock the anti-cancer drug, Imatinib, which is a small molecule kinase inhibitor, to the target receptor c-Abl kinase.
Software needed to complete this tutorial:
AutoDock Vina
MGLTools
PyMol
Open Babel
iDock
[Smina]
Background
Computational docking is used to perform in silico screening assays of possible therapeutic drugs. Most molecular docking methods make simplifications in order to make the computation tractable. One major simplification is that the receptor is treated as rigid so conformational flexibility of the active site is not taken into account. Because of these approximations, docking methods are useful for screening lots of compounds; however, other methods such as molecular dynamics should be used to refine predictions if a more realistic conformational search or energy prediction is needed. Molecular dynamics and related methods are complementary with computational docking methods.
AutoDock Vina is a fast and effective program for performing docking experiments. It is freely available here. AutoDock Vina uses a simple scoring function and rapid gradient-optimization conformational search. The scoring function is highly approximate, but is effective for typical drug-like ligands.
Note for those of you familiar with AutoDock: Unlike AutoDock, Vina does not require you to pre-calculate the grid map files. Vina calculates its grid maps automatically during the calculation.
Preparing the Protein Target
The RCSB pdb code for the target receptor c-Abl is 1IEP.pdb Before we can perform the docking experiment, we need to prepare the input crystal structure. We need to remove all duplicate and non-binding chains, all solvent molecules, salts, and non-binding metals and cofactors that are not next to the binding area. This can be readily accomplished in PyMol. Load the pdb file 1iep.pdb into PyMol. You will see two chains, several small ions, and water molecules that need to be removed. First remove chain B, the solvent molecules, and chloride ions by typing in the command line:
remove Chain B
remove solvent
remove name CLThe crystal structure of the inhibitor bound in the active site will be used as a test of our docking procedure. Select residue STI and save the residue as a pdb:
save drug_reference.pdb, resname STIWe need to determine the coordinates and dimensions of our search space. The easiest way to do this is to use the crystal structure with a ligand already bound in the pocket of interest. Select the molecule of interest and determine the center of mass:
select resname STI
centerofmass (sele)Here we see the center of mass coordinates in x, y, and z:
Center of Mass: [15.601, 53.387, 15.453]Write down these coordinates for later use.
Now we can remove the inhibitor and save the protein as the target for our docking experiment.
remove resname STI
save target.pdb, Chain AWe now have the pdb file, target.pdb, which contains just the protein chain A, to be used in our docking experiment.
Obtaining the Ligand
The structure of the drug molecule can be found at the DrugBank. Go to the website and read the description about Imatinib. Next to the Structure row, you will see the option to Download the structure. One option would be to download the structure as a pdb file and add hydrogens in a molecular editor such as Avogadro.
In this tutorial we will instead generate the geometry from a SMILES string, a text string that is a common chemical database format. We can use OpenBabel to attempt to generate the 3D geometry from the SMILES string.
The SMILES string can be found on the DrugBank site below the IUPAC Name. For Imatinib the SMILES string is
CN1CCN(CC2=CC=C(C=C2)C(=O)NC2=CC(NC3=NC=CC(=N3)C3=CN=CC=C3)=C(C)C=C2)CC1Copy this string to a text file called Imatinib.smi. Then we can generate a 3D conformation using OpenBabel by typing in the terminal:
obabel Imatinib.smi -O Imatinib.pdb --gen3d --conformer --nconf 50 --score energy The above command will run a conformer search to find the lowest energy structure and add hydrogens. The output file, Imatinib.pdb will be the lowest energy 3D conformer with hydrogens added. The structure is shown here:
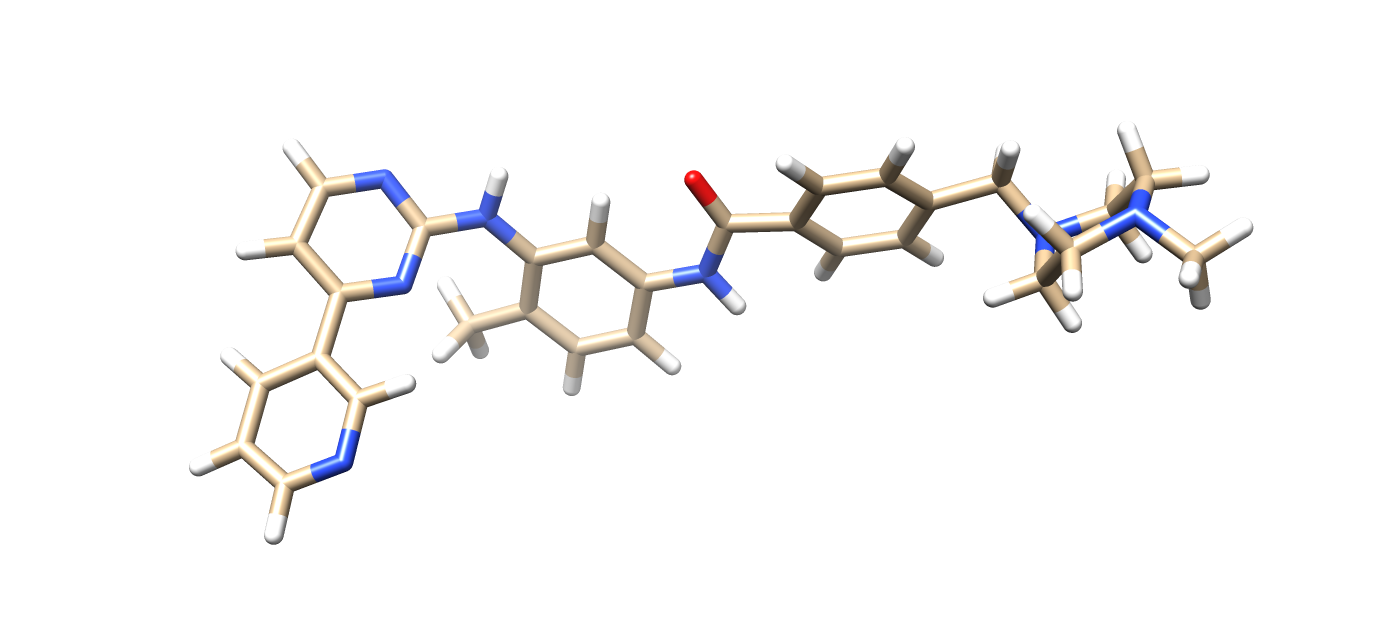
Molecular Docking with AutoDock Vina Using AutoDock Tools
Before we can use AutoDock Vina, we need to prepare the molecules for docking by adding charges, polar hydrogens, and converting the pdb file into pdbqt format. One way to do this using a graphical user interface is with the AutoDock Tools suite provided with MGLTools from the Center For Computational Structural Biology here
In this section we will use AutoDock Tools to prepare the protein and ligand for docking. If AutoDock tools is installed and in your PATH, then it can be loaded from the terminal with:
adtFirst we will load the protein file target.pdb prepared above by selecting File –> Read Molecule.
Since the pdb structure does not contain hydrogens we need to add them. Select Edit –> Hydrogens –> Add and choose the option Polar Only as shown:
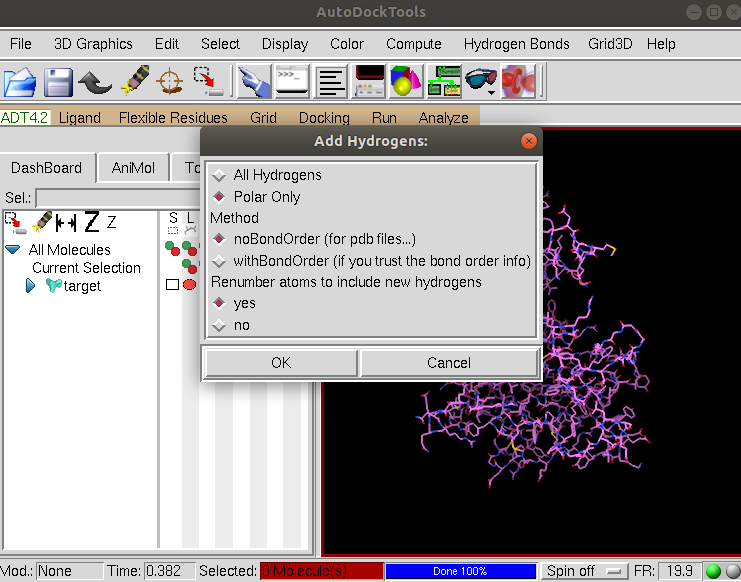
AutoDockTools will add all the polar hydrogens. Now we will save the structure in pdbqt format. Click on the Grid tab shown here:
And select Grid –> Macromolecules –> Choose and click on target and then the Select Molecule button:
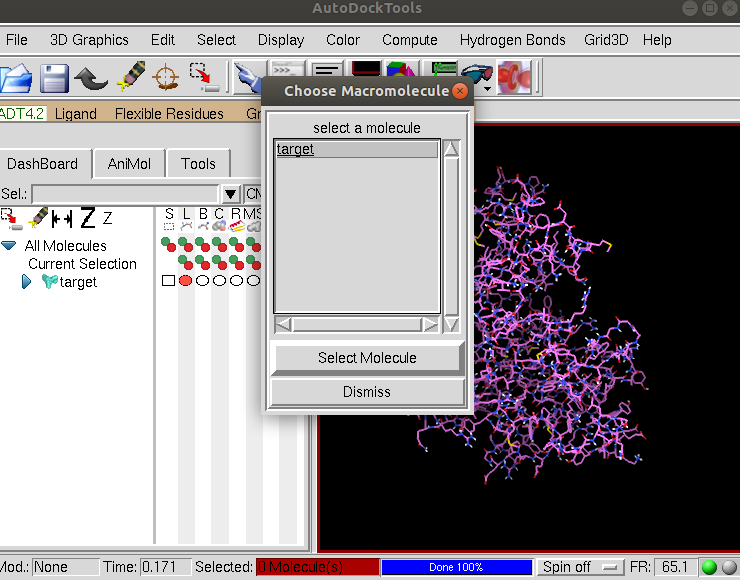
Click OK to continue and then Save to save the molecule in pdbqt format as target.pdbqt.
Now we need to prepare the Ligand. Select the Ligand tab as shown below:
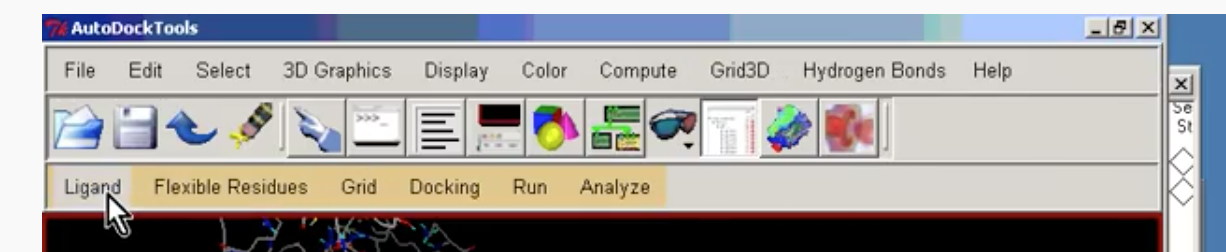
Select Ligand –> Input –> Open and change the Files of type to be PDB files as shown:
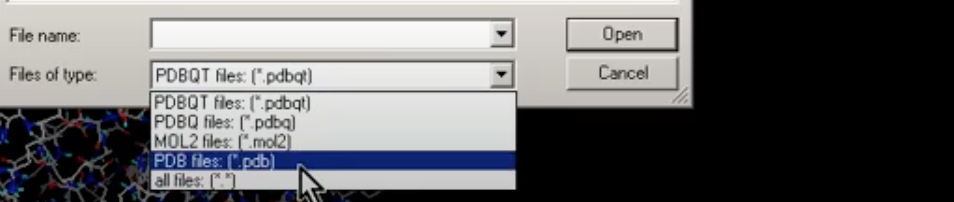
and select Imatinib.pdb and click Open. You will see summary for drug box as shown:
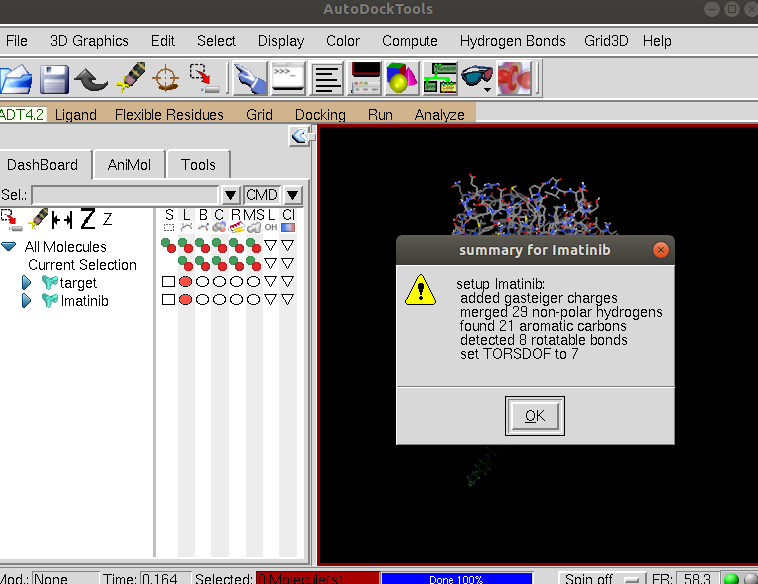
Click OK. We can hide the protein so as to see the ligand better by clicking on the red circle in the DashBoard on the left next to the target. Then re-center the view on the ligand by clicking on the Reset view icon. We see that the nonpolar hydrogens have been removed but the polar hydrogens remain:
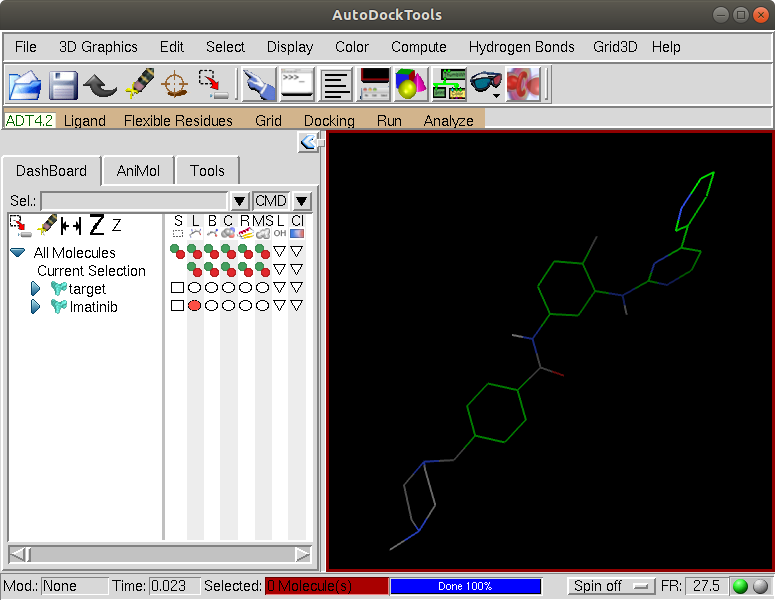
Now, we will examine the rotatable bonds in the ligand. Click on the tab Ligand –> Torsion Tree –> Choose Torsions
AutodockTools colors the rotatable bonds in green and the non-rotatable bonds in magenta. You can choose to make a bond rotatable by clicking on it. In this case, you should have seven rotatable bonds as shown in green:
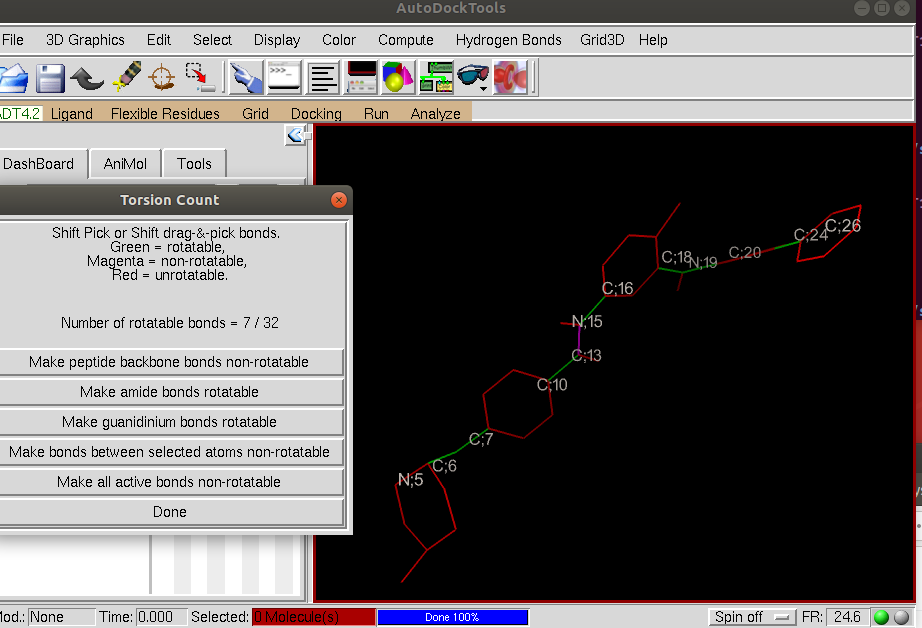
After inspecting the rotatable bonds, click Done and save the ligand structure by clicking on the tab Ligand –> Output –> Save as PDBQT and save as Imatinib.pdbqt
We are now done with AutodockTools, and you can close the adt graphical interface. We now have the files required for AutoDock Vina. These are the protein target file target.pdbqt and the drug file Imatinib.pdbqt.
To run Vina, we first need a properly formatted conf.txt file. An example is shown here:
receptor = target.pdbqt
center_x = 15.601
center_y = 53.387
center_z = 15.453
size_x = 20
size_y = 20
size_z = 20
out = output.pdbqt
exhaustiveness = 10
The first line in this script is the name of the target pdbqt file that contains the protein structure (target.pdbqt). We then specify the center of the search grid using the center of mass coordinates x, y, and z coordinates from our center of mass calculation above. Next, the size of the search space is specified by size_x, size_y, and size_z. Generally, a search space of 20x20x20 Angstroms is sufficient to accommodate drug-like molecules. The output file is specified to be output.pdbqt. Finally we set the exhaustiveness to 10. The exhaustiveness determines how exhaustively the search algorithm looks for the global minimum in energy of possible binding modes. A larger value of the exhaustiveness parameter means that the algorithm will be less likely to fail to find the global minimum, but the amount of time the calculation takes will increase with the exhaustiveness parameter.
Now run Vina by entering the following command:
vina --config conf.txt --ligand Imatinib.pdbqt --log log.txt The –ligand flag signals the name of the ligand pdbqt file. The results of the calculation will be written to the log file.
At the end of the calculation you should see something like:
mode | affinity | dist from best mode
| (kcal/mol) | rmsd l.b. | rmsd u.b
-----+-------------+-----------+---------
1 -12.9 0.000 0.000
2 -10.1 1.349 2.413
Writing output ... done.The output file output.pdbqt contains the structure of the binding pose configurations in pdbqt format. We can convert this to pdb format using Babel
babel -ipdbqt output.pdbqt -opdb output.pdb The output configurations can be visualized in Pymol, and you can scroll through the different configurations using the arrows on the bottom right. Here is an example of the lowest energy binding pose found by Autodock Vina compared with the ligand from the crystal structure shown in red.
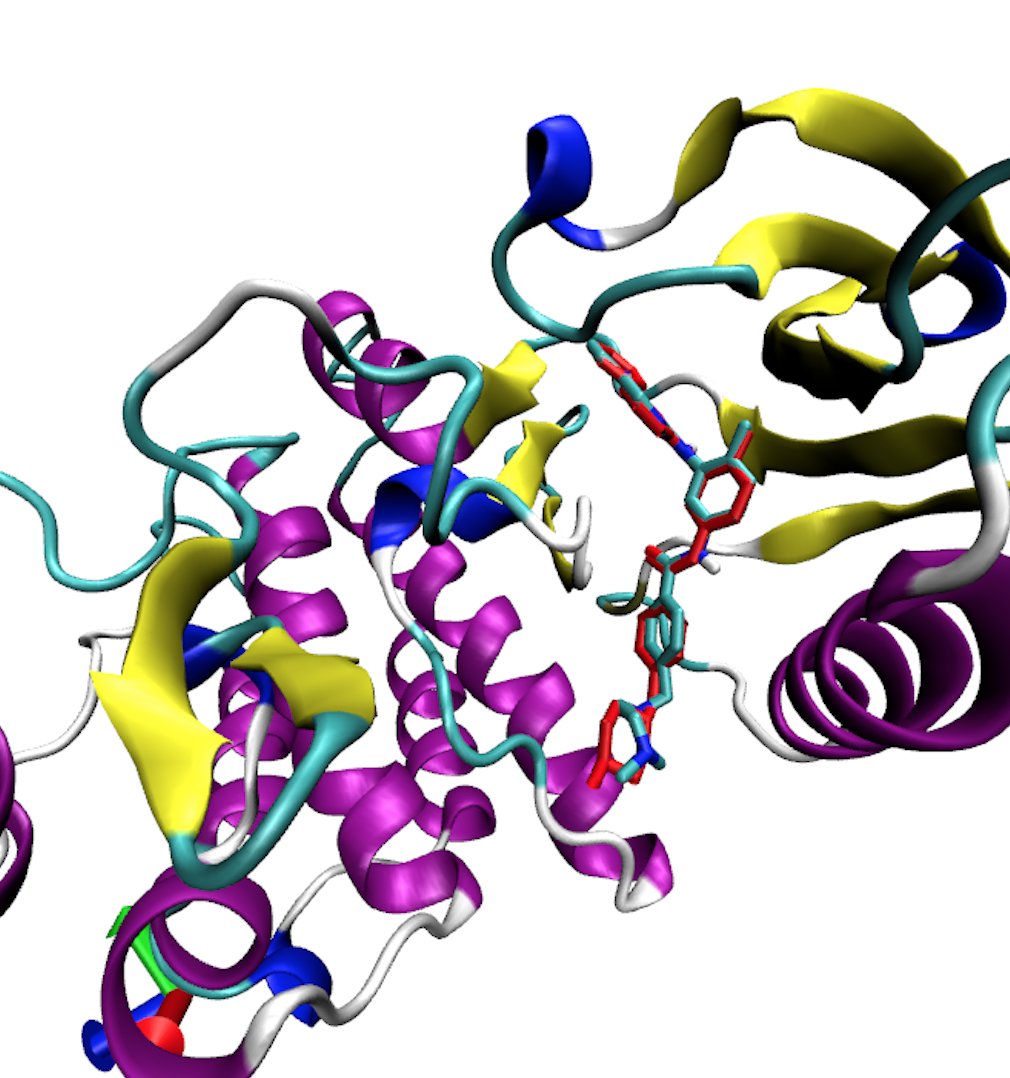
As we can see, Autodock Vina does a very good job at finding the binding pose in this case.
Molecular Docking with AutoDock Vina Using Python Scripts
Instead of using the graphical user interface of AutoDockTools, we can prepare our input files for Vina using the python scripts provided with MGLTools. These are located in MGLToolsPckgs/AutoDockTools/Utilities24/
Form the target pdb file that contains Chain A of our protein above, we can convert this to pdbqt format using the python script prepare_receptor4.py
prepare_receptor4.py -r target.pdb -A hydrogens -o target.pdbqtHere the -A flag signals to add hydrogens. The output will be target.pdbqt which has the receptor target in pdbqt format that can be used in Vina.
Next we need to prepare the ligand. We can use the python script prepare_ligand4.py and the ligand pdb file we created above called Imatinib.pdb
prepare_ligand4.py -l Imatinib.pdb -o Imatinib.pdbqt We now have our ligand in pdbqt format. Now we run Vina as before
vina --config conf.txt --ligand Imatinib.pdbqt --log log.txt At the end of the calculation you should see something like:
mode | affinity | dist from best mode
| (kcal/mol) | rmsd l.b. | rmsd u.b
-----+-------------+-----------+---------
1 -12.9 0.000 0.000
2 -9.9 1.232 2.066
Writing output ... done.As in the above example, convert this to pdb format using Babel
babel -ipdbqt output.pdbqt -opdb output.pdb Molecular Docking with iDock
iDock is another program that uses a similar scoring function as Vina. iDock has been optimized to run very fast, making it useful for virtual screening of large data bases. It is also useful to compare your results from Vina with iDock to get a consensus between different software. Using iDock is very similar to running AutoDock Vina: we need a target receptor and ligand in pdbqt format as above. To run iDock we need a properly formatted idock.conf file. An example is shown here:
receptor = target.pdbqt
out = ./Results
center_x = 15.601
center_y = 53.387
center_z = 15.453
size_x = 20
size_y = 20
size_z = 20
threads = 6
conformations = 10The file is similar to the one used for AutoDock Vina with a few small differences. We set the number of configurations to be written to the output pdbqt folder to 10. The output pqdqt and log.csv file containing information of the iDock score will be written to a new directory called Results. To run iDock we type the command:
idock --config idock.conf --ligand Imatinib.pdbqt
At the end of the calculation you should see something like:
Index Ligand nConfs idock score (kcal/mol) RF-score (pKd)
1 Imatinib 10 -13.03 7.52and the output pdbqt structure will be written to the directory Results.
Virtual Screening
Docking calculations can be useful to predict the binding affinity of proposed ligands prior to organic synthesis in order to screen a large number of candidate molecules against a biochemical target. In this exercise we demonstrate this capability by screening several cyclin-dependent kinase type-2 (CDK2) inhibitors. As an example, we will use the indazole series from J. Chem. Edu. 2017, 94, 345-349
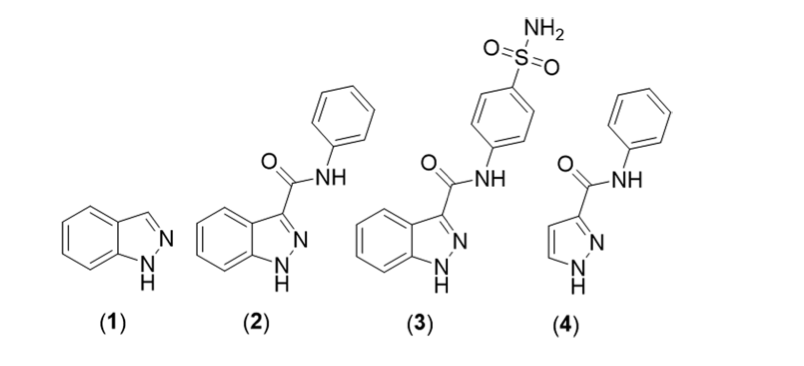
Structures of CDK2 inhibitors.
To run our screening calculation we need
- A directory named Ligands that contains all of the input structures in PDBQT format. An example can be found here.
- The target protein file in PDBQT format. In this case we will use the CDK5 target from pdb entry [2VTA] available here.
- A proper Vina configuration [file] or iDock configuration file.
An example Vina configuration file is shown below:
receptor = ../target.pdbqt
center_x = 27.86
center_y = 0.69
center_z = 66.23
size_x = 20
size_y = 20
size_z = 20
out = output.pdbqt
num_modes = 10
exhaustiveness = 80In this case we set the exhaustiveness to 80 and write 10 final binding configurations. The bash script screen_vina.bash will perform the docking calculation for each compound in the Ligands directory and write the output to the Results-Vina directory:
bash screen_vina.bashA similar screening procedure can be performed using iDock with the iDock configuration file shown below:
receptor = target.pdbqt
ligand = ./Ligands
out = ./Results-iDock
center_x = 27.86
center_y = 0.69
center_z = 66.23
size_x = 20
size_y = 20
size_z = 20
threads = 6
trees = 5000
tasks = 640
conformations = 10We set trees = 5000 and tasks = 640 to do a more exhaustive screening and write 10 final binding configurations. The bash script screen_idock.bash will perform the docking calculation with iDock for each compound in the Ligands directory and write the output to the Results-iDock directory:
bash screen_idock.bashA comparison between the docking score and the experimentally determined IC50 is shown below:
Compound actual affinity (IC50) Vina predicted affinity (kcal/mol) iDock predicted affinity (kcal/mol) iDock Kd (uM)
1 185 -5.6 -5.65 363
2 3 -8.1 -8.27 0.32
3 0.66 -8.7 -8.20 1.0
4 97 -6.7 -6.81 16.59As we can see the docking score generated by autodock Vina correctly predicts that compound 3 will have the highest binding affinity (most negative predicted free energy score) and compound 1 will have the lowest (least negative predicted affinity). iDock predicts a similar trend but predicts that Compounds 2 and 3 will have similar affinities.