Replica Exchange Molecular Dynamics (REMD)
Running and analyzing multi-replica simulations in GROMACS with PLUMED2
About this tutorial
In this tutorial we will use GROMACS patched with PLUMED2 to run simulations with multiple replicas and analyze them. In particular, we will focus on parallel tempering (PT) where the replicas are run at different temperatures. This tutorial was created using GROMACS-2019.4 and PLUMED2.7.0 compiled with mpi. As an example we will set up a REMD simulation of alanine dipeptide in vacuum and calculate free energies from the simulation. This tutorial is based on the Belfast tutorial as part of the suite of PLUMED2 tutorials.
Summary of theory
In replica exchange molecular dynamics (REMD), several non-interacting replicas of the same system are run in parallel according to a different Hamiltonian. At fixed intervals, an exchange of configurations between two replicas is attempted. A popular case of REMD is parallel tempering (PT), in which different replicas of the system are simulated using the same potential energy function, but at increasing temperatures in order to accelerate sampling of phase space. The basic idea is that the high temperature replicas will sample phase space more efficienty due to the increased thermal energy, which these replicas from getting trapped in local minima. In a PT simulation, exchanges are periodically attempted between replicas at adjacent temperatures according to the following acceptance probability:
\[\begin{equation} \label{EQ:1} p(i \rightarrow j) = min\{ 1,e^{\Delta^{PT}_{i,j}}\}\end{equation}\]with
\[\begin{equation}\label{EQ:2} \Delta^{PT}_{i,j} =\left( \frac{1}{k_BT_i}-\frac{1}{k_BT_j}\right)\left(U(R_i)-U(R_j)\right) \end{equation}\]where \(R_i\) and \(R_j\) are the configurations at temperature \(T_i\) and \(T_j\), respectively.
The acceptance probability is determined by the overlap between the energy distributions of two replicas. An efficient diffusion in temperature space is necessary to to ensure an adequate configurational sampling, and this in turn requires an overlap between the energy distributions of two replicas. The number of temperatures needed to cover a given temperature range scales as the square root of the number of degrees of freedom, making this approach prohibitively expensive for large systems.
Setup and run a PT simulation
We will run a PT simulation of alanine dipeptide in vacuum, using 4 replicas at the following temperatures: 300K, 400K, 600K, and 1000K. To examine conformational changes, we will monitor the time evolution of the two dihedral angles phi and psi using PLUMED2. First we need the following plumed.dat file that will be fed into PLUMED while using 4 replicas
#SETTINGS NREPLICAS=4
# Set up two variables for Phi and Psi dihedral angles
phi: TORSION ATOMS=5,7,9,15
psi: TORSION ATOMS=7,9,15,17
PRINT STRIDE=10 ARG=phi,psi FILE=COLVARNow create 4 directories sim0, sim1, sim2, and sim3 where each individual replica will be run
mkdir sim0 sim1 sim2 sim3The GROMACS topology (.top), structure file (.gro), parameter file (.mdp) can be found here. Each of these files need to be copied to each of the replica directories. In the .mdp file, we need to replace __T__ with the temperature for that replica. The bash script gentemps.sh will copy the files and update the reference temperatures. Copy the files alanine_dipeptide.gro, topol.top, vacuum.mdp, and gentemps.sh to the same directory and run
bash gentemps.shIf you want to check the reference temperatures for the thermostats in each, you can do that efficiently with a command like
grep ref_t sim*/vacuum.mdpNow we want to run grompp in each directory to build the independent .tpr files we need. We could cd to each directory in turn and run grompp, or you could use a bash shell to write a quick loop to do that operation:
(for dir in sim[0123]; do cd $dir; gmx_mpi grompp -f vacuum.mdp -c alanine_dipeptide.gro -p topol.top; cd ..; done)This says to loop over sim0 … sim3, and for each of those, change into the directory, run grompp and then change back to the parent directory.
Now we are ready to perform the replica exchange MD. To submit this simulation with Gromacs type
mpirun -np 4 gmx_mpi mdrun -v -plumed ../plumed.dat -multidir sim[0123] -replex 100This command will execute 4 MPI processes in parallel using the files topol0.tpr … topol3.tpr stored in the sim0 … sim3 sub-directories. An exchange between configurations will be attempted every 100 steps. The output files produced by PLUMED will be renamed and a suffix indicating the replica id will be appended.
Analyzing a REMD simulation
We will start by inspecting the output file COLVAR.0, which reports the time evolution of the CVs at 300K. The COLVAR.0 file wil be located in the sim0 directory. We can construct a histogram of the CVs using the HISTOGRAM command as part of the plumed analysis module. See the Building Histograms in PLUMED tutorial. In this case we will make a new plumed file plumed-histo.dat with the following lines
rphi: READ FILE=COLVAR.0 VALUES=phi INGORE_TIME
rpsi: READ FILE=COLVAR.0 VALUES=psi IGNORE_TIME
HISTOGRAM ...
ARG=rphi,rpsi
STRIDE=1
GRID_MIN=-pi,-pi GRID_MAX=pi,pi GRID_BIN=200,200
KERNEL=DISCRETE
LABEL=hh
... HISTOGRAM
ff: CONVERT_TO_FES GRID=hh TEMP=300
DUMPGRID GRID=ff FILE=fes.datHere we are building a discrete 2D histogram of the sample phi and psi angles and then outputting the corresponding free energy surface called fes.dat Run the plumed driver by typing
plumed driver --noatoms --plumed plumed-histo.datThis will output an estimate of the free energy surface in the file called fes.dat. An example plot of the free energy surface is shown below:
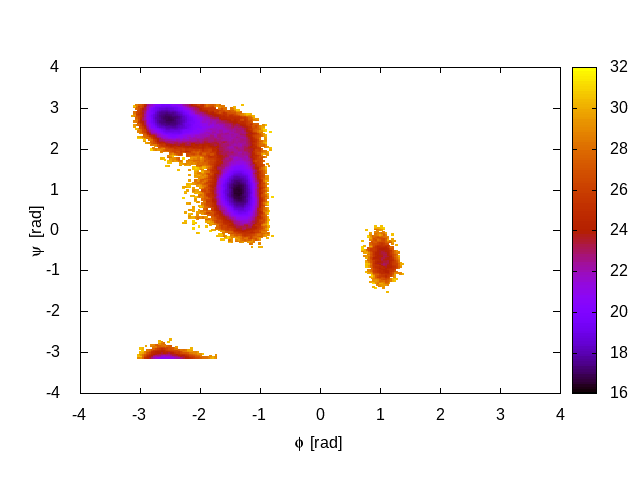
In PT simulations we also have to examine the time evolution of each replica diffusing in temperature space, to make sure that we are all the replicas are accessing the highest temperature and that all the replicas are exploring all the relevant basins. To do this we use the pearl script provided with GROMACS demux.pl To demux the trajectories type the following command:
demux.pl sim0/md.logThis will create two files, called replica_temp.xvg and replica_index.xvg. A plot of replica_temp.xvg is shown below:
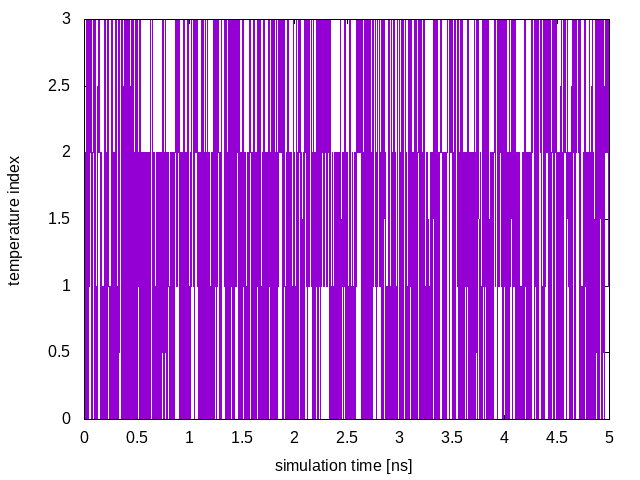
This plot shows the temperature index of each configuration at a given time of the simulation and allows us to monitor each replica’s diffusion in temperature. In order for the PT algorithm to be effective, we need an efficient diffusion of the replicas in temperature space. In the above plot we see all replicas are diffusing effectively in temperature.
Next, we need to monitor the phase space sampled by each replica while diffusing in temperature and verify that each replica is interconverting between the different basins of the free-energy landscape. We use the replica_index.xvg file to reconstruct continuous trajectories of each replica in temperature using the trjcat command:
gmx_mpi trjcat -f sim0/traj.trr sim1/traj.trr sim2/traj.trr sim3/traj.trr -demux replica_index.xvg -o 0_trajout.xtc 1_trajout.xtc 2_trajout.xtc 3_trajout.xtcTo analyze the demuxed trajectories we can use the PLUMED driver again. Make a new plumed file called plumed_demux.dat with the following:
# Set up two variables for Phi and Psi dihedral angles
phi: TORSION ATOMS=5,7,9,15
psi: TORSION ATOMS=7,9,15,17
PRINT STRIDE=1 ARG=phi,psi FILE=COLVAR-demuxand run the driver with the command
plumed driver --mf_xtc 0_traj.xtc --plumed plumed_demux.datand rename the output:
mv COLVAR-demux COLVAR-demux.0Repeat this procedure for the other replicas, renaming the COLVAR-demux to COLVAR-demux.1, COLVAR-demux COLVAR-demux.2, and COLVAR-demux COLVAR-demux.3
Below is a plot of the sampled configurations for the first two replicas. It is clear that in this case replicas are able to interconvert between the two metastable states
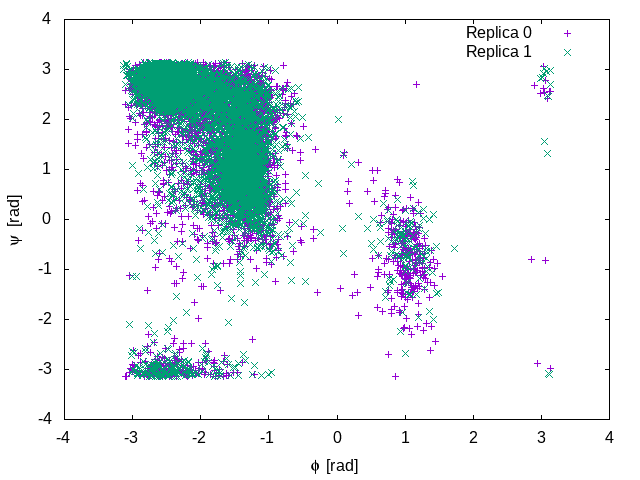
The Well-Tempered Ensemble (PT-WTE simulation)
In this tutorial we will combine PT replica exchange with metadynamics in the well-tempered ensembled. In the well-tempered ensemble we bias the system’s potential energy using metadynamics. Parallel tempering in its well-tempered ensemble version (PT-WTE), has been used to study loop conformational polymorphism in cellular prion proteins.
We will perform a PT-WTE simulation of alanine dipeptide in water. The GROMACS topology (.top), structure file (.gro), parameter file (.mdp) can be found here
First, we run a short PT simulation using 4 replicas at temperatures between 300K and 400K. We will use a geometric distribution of temperatures, thus our 4 replicas will be run at T=300, 330.2, 363.4 and 400K. In this equilibration stage we can monitor the two dihedral angles and the total energy of the system with the PLUMED file:
#SETTINGS NREPLICAS=4
# Set up three variables for Phi and Psi dihedral angles and total energy
phi: TORSION ATOMS=5,7,9,15
psi: TORSION ATOMS=7,9,15,17
ene: energy
PRINT STRIDE=10 ARG=phi,psi,ene FILE=COLVAR_PTThe bash script build_topo.sh will copy the files, compute the temperature from the geometric distribution, build the independent .tpr files using the grompp command. Copy the files alanine_dipeptide_water.gro, topol.top, TEMPLATE_md.mdp, and build_topo.sh to the same directory and run
bash build_topo.shAnd run the simulations as before:
mpirun -np 4 gmx_mpi mdrun -v -plumed ../plumed.dat -multidir sim[0123] -replex 100At the end of the run, we need to analyze the acceptance rate between exchanges. This quantity is reported at the end of the Gromacs output file, called md.log, and it can be extracted using the following bash command line:
grep -A2 "Repl average probabilities" sim0/md.logIn this case, the output reads:
Repl average probabilities:
Repl 0 1 2 3
Repl .00 .00 .00From the line above, we see that none of the attempted exchanges has been accepted. The reason is that we did not choose enough replicas to cover the temperature range 300-400K. In the current setup there is a poor overlap of the potential energy distributions at different temperatures. We can see this by plotting the total energy in each replica as a function of time:
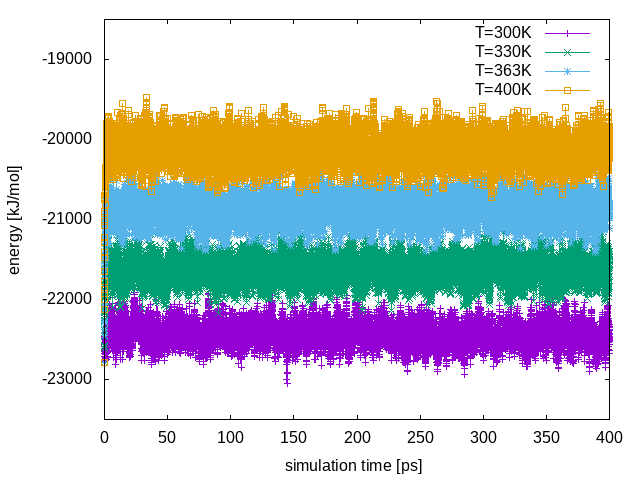
In order to see exchanges between the different replicas, we need to improve the overlap of the potential energy distributions at the different temperatures. The conventional way to achieve this is to increase the number of replicas; however, this will increase the computational cost. Instead, we will enlarge the fluctuations of the energy by sampling the Well-Tempered Ensemble (WTE). To do this, we need to setup a well-tempered metadynamics simulation using energy as the collective variable (CV). In WTE, fluctuations of the energy will be enhanced by a factor equal to the square root of the biasfactor. In this example, we will enhance fluctuations of a factor of 4, thus we will set the biasfactor equal to 16.
In PLUMED2.4 or later, we can use a simple syntax to manipulate multiple replica inputs with only small differences in the input files. A sample plumed file is shown here:
#SETTINGS NREPLICAS=4
# Set up three variables for Phi and Psi dihedral angles and total energy
phi: TORSION ATOMS=5,7,9,15
psi: TORSION ATOMS=7,9,15,17
ene: energy
METAD ...
ARG=ene
PACE=250
HEIGHT=1.2 SIGMA=140.0
FILE=HILLS_PTWTE
BIASFACTOR=16.0
TEMP=@replicas:300.0,330.2,363.4,400
LABEL=metad
... METAD
PRINT STRIDE=10 ARG=phi,psi,ene,metad.bias FILE=COLVAR_PTThe @replicas keyword changes the temperature parameter in the plumed.dat file for each replica. In the above file we are signaling to perform metadynamics using the METAD directive.
We specify the Gaussian width, SIGMA=140.0 to be on the order of the fluctuations of the potential energy at 300K as calculated from the preliminary PT run and seen in the figure above.
We run the simulation following the usual procedure:
mpirun -np 4 gmx_mpi mdrun -v -plumed ../plumed.dat -multidir sim[0123] -replex 100Now, when we analyze the average probability acceptance with:
grep -A2 "Repl average probabilities" sim0/md.logwe notice that now on average 24% of the exchanges are accepted.
Repl average probabilities:
Repl 0 1 2 3
Repl .23 .24 .25Now demux the trajectories using the pearl script demux.pl as above:
demux.pl sim0/md.logA plot of replica_temp.xvg is shown below:
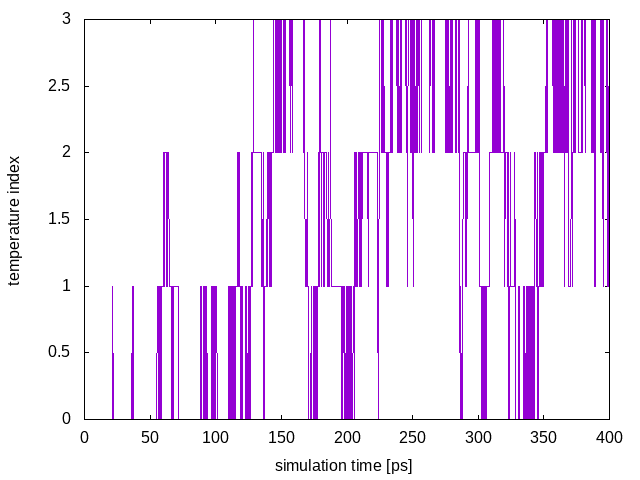
Here we see that the system is efficiently diffusing in the entire temperature range and no bottlenecks are present. As was done in the PT simulation, we can look at the energy collective variable as a function of time for each replica:
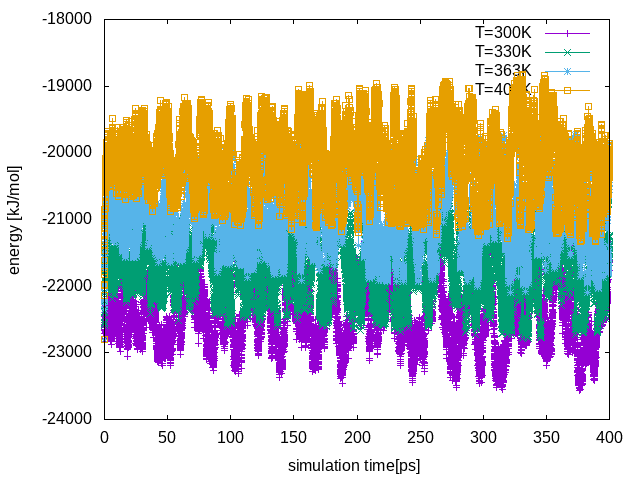
If we compare this plot with the one from the PT run, we notice that the fluctuations in energy are enhanced due to the metadynamics bias, leading to a better overlap between energy distributions at different temperatures. This increased overlap in energy increases the exchange probability in our replica exchange.
In order to obtain an estimate of the free energy, we need to correctly account for the metadynamics bias.