Geometry Optimization
About this tutorial
In this tutorial we will perform a geometry optimization using various QM simulation software packages. We will perform a geometry optimization and single point energy calculation for the molecule 1-methylcylohexene using GAMESS, Orca, and CP2K QM software.
Software needed to complete this tutorial:
Getting Initial Structure
Obtain the initial structure for 1-methylcylohexene. One way to get an initial structure is to sketch the chemical structure of the molecule using the online JME Editor.
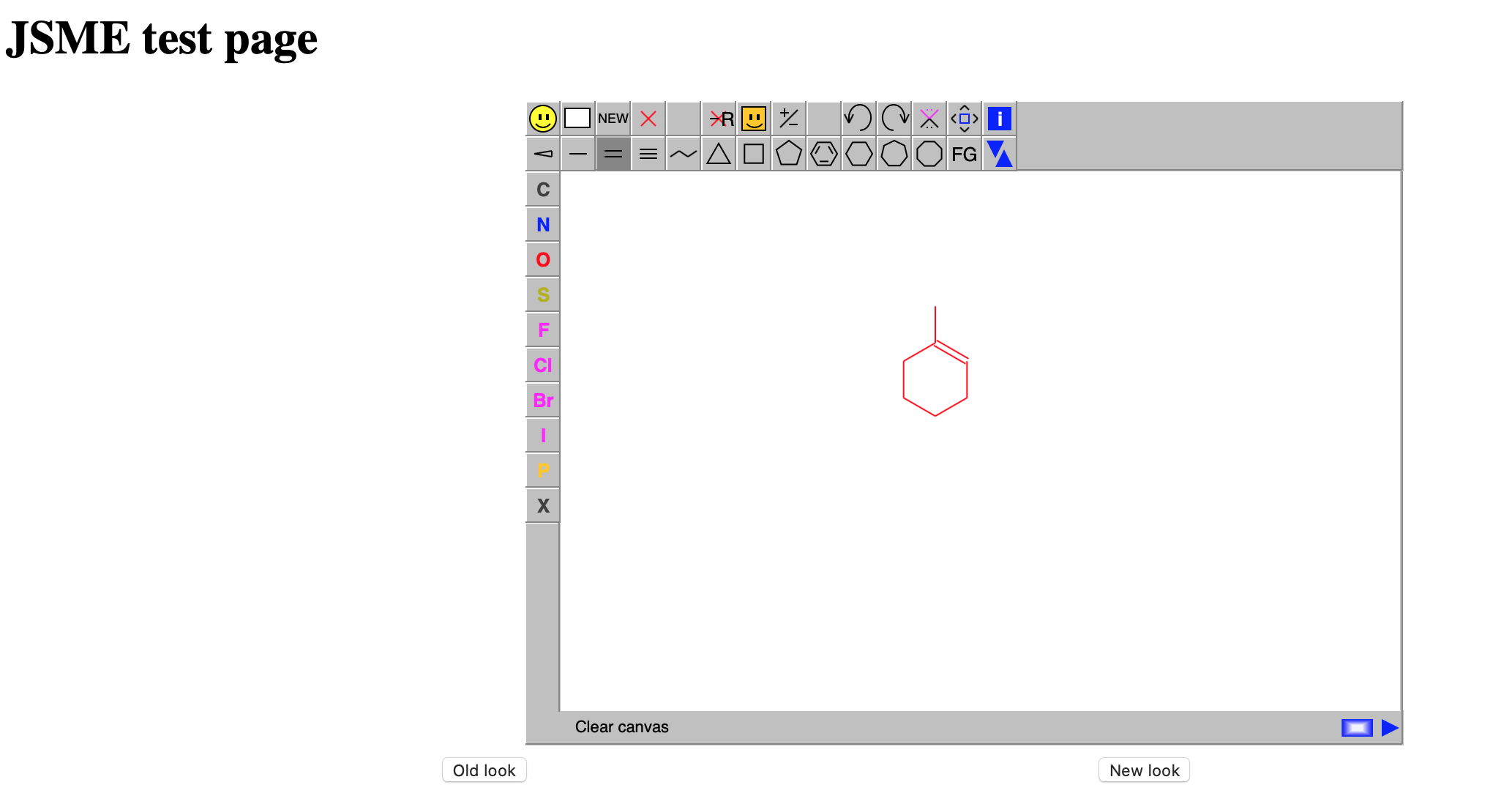
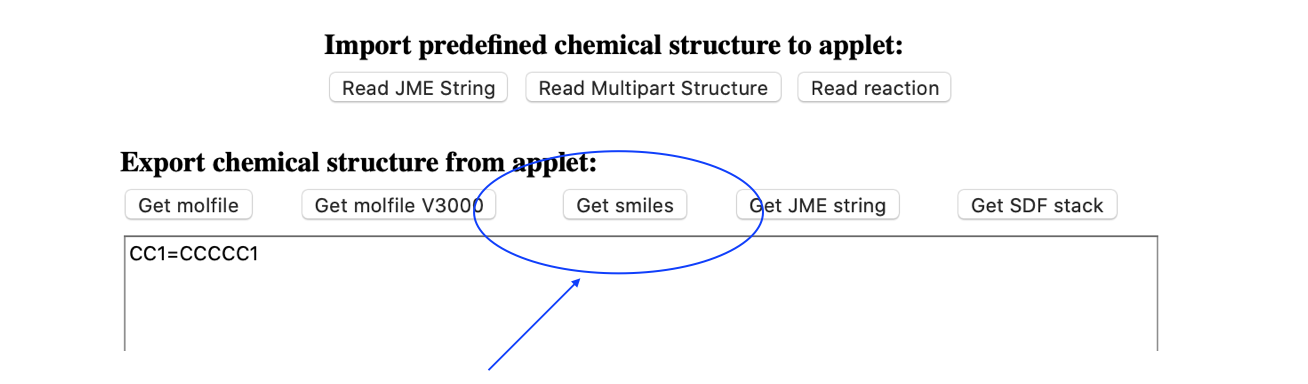
You can then get the SMILES format and save this to a text file called 1-methylcyclohexene.smi
CC1=CCCCC1We can then use Open Babel to convert the SMILES string into a 3D structure that we will save in pdb format:
obabel 1-methylcyclohexene.smi -O 1-methylcyclohexene.pdb --gen3d --conformer --nconf 50 --score energyand we can convert this pdb file into a GAMESS input file: convert to input file
obabel -i pdb 1-methylcyclohexene.pdb -o inp -O 1-methylcyclohexene.inpGeometry Optimization in GAMESS
In this section we will optimize the geometry of the 1-methylcyclohexene molecule using the B3LPY DFT functional and 6-31+G(d,p) basis set using GAMESS.
Here the $CONTRL group indicates SCFTYPE=RHF for a closed-shell molecule, RUNTYPE=OPTIMIZE for a geometry optimization, ICHARG=0 specifies the charge is 0, and MULT=1 specifies the multiplicity. NPRINT=-5 specifies that only minimal output is produced. The DFT type is also specified in the $CONTRL section. With DFTTYP=NONE (default), an ab initio (Hartree Fock) calculation will be performed rather than density functional theory. In this case we will use DFTTYPE=B3LYP to select for the B3LYP hybrid method.
We specify the basis set with the $BASIS group section in which we define N-31G type basis by GBASIS=N31 with the N in N-31G type basis set by NGAUSS=6. We also define a single (d) function on heavy atoms by NDFUNC=1, a single (p) function on hydrogens by NPFUNC=1, and a single (+) diffuse sp shell on heavy atoms by DIFFSP=.TRUE. Finally, we specify to use the DFT-D3(BJ) dispersion correction with $DFT IDCVER=4.
The input file to run the geometry optimization should look like this:
$CONTRL SCFTYP=RHF MULT=1 ICHARG=0 RUNTYP=OPTIMIZE
DFTTYP=B3LYP MAXIT=60 NPRINT=-5 $END
$BASIS GBASIS=N31 NGAUSS=6 NDFUNC=1 NPFUNC=1 DIFFSP=.T. DIFFS=.F. $END
$SYSTEM MWORDS=200 $END
$SCF DIRSCF=.T. $END
$DFT IDCVER=4 $END
$DATA
1-methylcyclohexene
C1
C 6.0 2.51500000 0.05200000 0.06800000
C 6.0 1.01500000 0.06100000 0.00700000
C 6.0 0.30900000 1.20200000 -0.08900000
C 6.0 -1.19100000 1.25100000 -0.10900000
C 6.0 -1.83700000 -0.05600000 0.35200000
C 6.0 -1.14500000 -1.25900000 -0.27800000
C 6.0 0.34600000 -1.29200000 0.06700000
H 1.0 2.85200000 -0.40800000 1.00300000
H 1.0 2.92600000 -0.52100000 -0.76900000
H 1.0 2.93600000 1.06200000 0.02200000
H 1.0 0.82200000 2.15900000 -0.14900000
H 1.0 -1.53800000 2.06700000 0.53500000
H 1.0 -1.51600000 1.48600000 -1.12900000
H 1.0 -1.77400000 -0.13100000 1.44600000
H 1.0 -2.90100000 -0.05400000 0.09400000
H 1.0 -1.62100000 -2.18700000 0.06000000
H 1.0 -1.26800000 -1.21800000 -1.36800000
H 1.0 0.84500000 -1.98400000 -0.62200000
H 1.0 0.47500000 -1.69600000 1.07900000
$ENDTo run the geometry optimization type:
rungms gamess.inp 01 4 >& geom_opt.logwhere 01 is the version number and 4 specifies to use 4 compute processes.
Basic usage of GAMESS is:
rungms [input] 01 [num procs] [num nodes] >& logGAMESS reports energy values in Hartree units (1 Hartree = 627.51 kcal/mol). You can use the Unix command grep to display relevant lines:
grep 'ENERGY= ' geom_opt.logFor 1-methylcyclohexene the final energy after 12 iterations is -273.8239986. To improve the accuracy we can choose a different basis set or a different DFT method using the optimized geometry as as the starting coordinates.
Geometry Optimization in CP2K
Geometry optimization can also be performed in CP2K. CP2K requires a basis set file.
Geometry Optimization in GAMESS using B3LYP/DEF2TZVP
python build-gamess-basis.py -inp 1-methylcyclohexene.inp -bas def2-tzvp.1.bas -o methyl_geom_opt.inpThe crystal structure of the inhibitor bound in the active site will be used as a test of our docking procedure. Select residue STI and save the residue as a pdb:
rungms 01 [input] [num procs] [num nodes] >& logGAMESS reports energy values in Hartree units (1 Hartree = 627.51 kcal/mol). You can use the Unix command grep to display relevant lines:
grep 'ENERGY= ' geom_opt.logGeometry Optimization in CP2K
Higher-level Single Point Energy
Copy the coordinates from the geometry optimization EQUILIBRIUM GEOMETRY LOCATED runtyp=energy mplevl=4 to enable MP4(SDQ) single point calculation
locate RESULTS OF MOLLER-PLESSET 4TH ORDER CORRECTION ARE